You need to enable JavaScript to run this app.
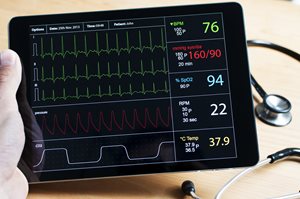
Is your software a medical device?
Feature Articles
Randolph Fillmore